研究人员解析阿尔茨海默病关键病理特征协同致病机制





文章导读
你可能听说过两个与阿尔茨海默病密切相关的“凶手”:大脑里异常聚集的Tau蛋白,以及持续下降的葡萄糖代谢。但它们之间的关系,长期以来就像两条平行线——各研究各的,没人说得清它们到底怎么合作作案。最近,一项来自中国科学院的研究终于揭开了这个谜底:原来当葡萄糖代谢走下坡路时,Tau蛋白会突然“黑化”,从缠结的“死结”变成主动招募死亡通路的“分子陷阱”。它会直接绑架一种叫RIPK1的关键激酶,解除细胞最后的生存防线,把神经元推向不可逆的坏死。
— 内容由好学术AI分析文章内容生成,仅供参考。
阿尔茨海默病中,神经细胞内磷酸化Tau蛋白(p-Tau)的异常聚集与神经退行性病变相关。同时,大脑葡萄糖代谢降低是预测认知功能障碍的重要临床指标。一个如同缠住神经元的“结”,一个好似掐断能量供给的“闸”,但这两个特征如何协同作用并共同驱动阿尔茨海默病进程,仍是尚未阐明的问题。
近日,中国科学院上海有机化学研究所等,揭示了葡萄糖代谢降低与p-Tau协同作用,通过调控RIPK1介导的程序性坏死通路,驱动阿尔茨海默病发生发展的分子机制。
体外细胞实验与体内动物模型研究表明,葡萄糖代谢降低与p-Tau并非独立发挥作用,而是形成协同效应,共同诱导神经元程序性坏死并导致神经元丢失。在低浓度葡萄糖环境下,异常积累的p-Tau可作为功能性分子支架,直接招募程序性坏死关键激酶RIPK1。同时,程序性坏死检查点蛋白A20表达下调,削弱其对该通路的负向调控作用,从而解除对细胞死亡的抑制。
这一“协同调控”机制共同促进神经元程序性坏死。在干预层面,补充乙酰左旋肉碱恢复A20表达,或利用RIPK1中间结构域来源的肽段,阻断p-Tau与RIPK1的相互作用,均可在Tau转基因小鼠模型中抑制神经元程序性坏死,减轻脑萎缩,并缓解学习记忆功能损伤。
该研究揭示了一条由p-Tau–RIPK1轴介导的代谢驱动型程序性坏死机制,为理解大脑葡萄糖代谢与Tau病理之间的功能联系提供了依据,并为阿尔茨海默病的干预策略提供了新思路。
相关研究成果在线发表在《神经元》(Neuron)上。研究工作得到国家自然科学基金委员会和中国科学院等的支持。
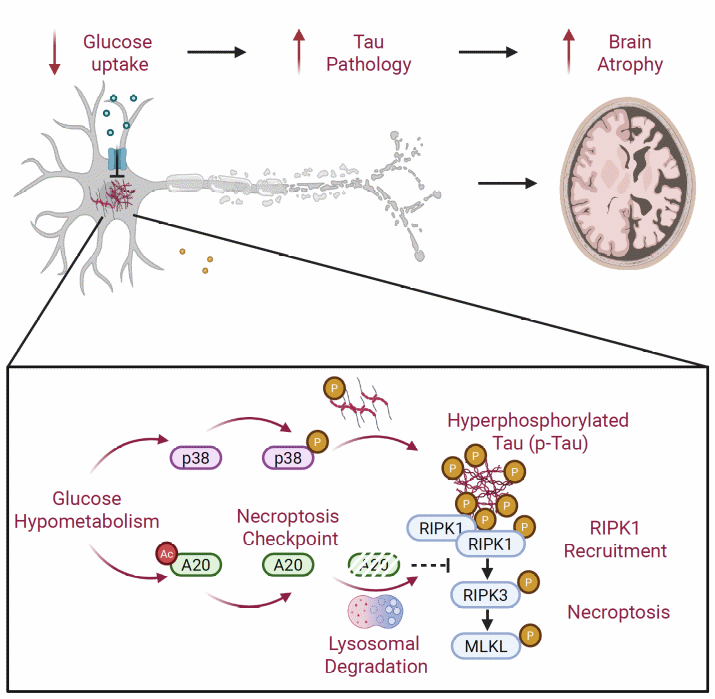
葡萄糖代谢降低与p-Tau协同驱动神经元程序性坏死的作用机制示意图
© 版权声明
本文由分享者转载或发布,内容仅供学习和交流,版权归原文作者所有。如有侵权,请留言联系更正或删除。





相关文章




这实验听起来挺玄的,脑子都晕了 😂