新研究发现调控神经轴突退化机制







文章导读
神经损伤后,为何轴突会不可逆崩解?中科院上海有机所方燕姗团队最新研究揭开关键谜底:FBXO21蛋白竟是加速神经退化的“隐形推手”!团队首次鉴定出该蛋白通过泛素化精准切割NMNAT2——这个维持轴突生命的“守护者”,导致损伤后神经快速瓦解。实验证实,敲除FBXO21后,小鼠神经组织中NMNAT2水平飙升,受损轴突退化速度延缓超50%。这项发表于《细胞生物学杂志》的突破,不仅破解了沃勒变性百年机制,更指向治疗脊髓损伤、阿尔茨海默病的全新靶点——阻断这一开关,或能让神经损伤修复迎来转机。
— 内容由好学术AI分析文章内容生成,仅供参考。
神经轴突损伤后,远离胞体的一侧发生渐进性串珠化、碎片化改变,进而崩解并被清除,这一病理过程被称为沃勒变性。NMNAT2是维持轴突完整性的关键蛋白,在神经损伤后快速耗竭而致沃勒变性发生,但在神经元中调控其蛋白降解的具体机制尚不完全清楚。
近日,中国科学院上海有机化学研究所方燕姗团队,鉴定出FBXO21是介导神经元中NMNAT2泛素化和降解的特异性F-box蛋白和关键因子。研究发现,在小鼠背根神经节神经元中,敲低Fbxo21引起NMNAT2蛋白水平升高,并延缓损伤引起的轴突退化。
时间动态分析证明,NMNAT2蛋白在神经元胞体和轴突中、损伤前后的降解速率基本相当,提示NMNAT2降解具有相同的调控机制,而受损轴突中NMNAT2的快速耗竭主要归因于缺少胞体来源的新生NMNAT2蛋白。无论是在胞体还是轴突、无论是完好还是受损的轴突,FBXO21对NMNAT2蛋白稳定性以及轴突完整性均发挥着重要的调控作用。
变性免疫沉淀和体外泛素化实验显示,FBXO21可与SKP1、CUL1和RBX1形成SCFFBXO21E3泛素连接酶复合物,介导NMNAT2的泛素化。SCFFBXO21泛素化NMNAT2中ISTID结构域的赖氨酸155(K155),该位点的泛素化缺陷突变K155R延长NMNAT2半衰期,具有更强的轴突保护作用。将NMNAT1或NMNAT3亚型中的ISTID域替换为NMNAT2的ISTID,导致原本稳定的前两者蛋白快速降解,而在其中引入K155R突变则可消除此效应。这说明,在进化上,NMNAT2的超短半衰期依赖于ISTID中的K155泛素化,揭示了NMNAT2独特的蛋白不稳定性的分子结构基础。
科研团队构建出Fbxo21敲除小鼠,并发现NMNAT2蛋白水平在该小鼠的神经组织中特异性升高。进而,团队利用小鼠坐骨神经损伤模型,证实了Fbxo21敲除可在体内延缓受损神经轴突的退化。
上述研究揭示了FBXO21是调控NMNAT2稳定性的关键因子,为轴突退化机制提供了新见解。同时,研究鉴定出SCF复合物中决定NMNAT2底物特异性的F-box蛋白FBXO21,为靶向NMNAT2治疗神经损伤和神经退化疾病提供了可能的靶点和干预策略。
相关研究成果在线发表在《细胞生物学杂志》(Journal of Cell Biology)上。研究工作得到国家自然科学基金、国家重点研发计划、中国科学院相关项目等的支持。
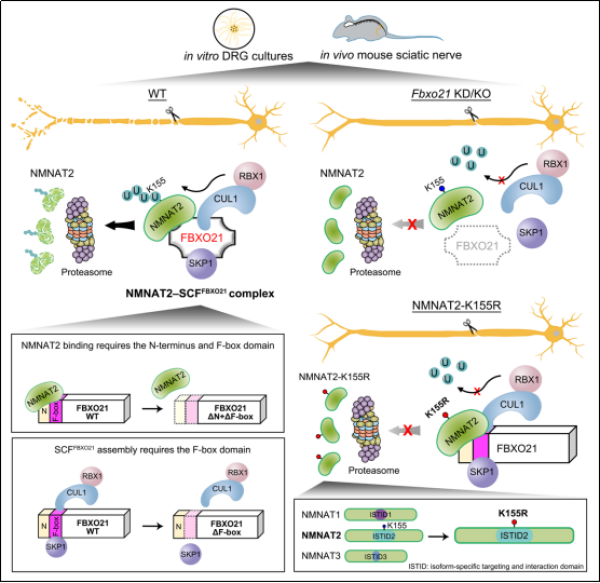
FBXO21介导NMNAT2泛素化和蛋白降解从而调控轴突退化的机制示意图
© 版权声明
本文由分享者转载或发布,内容仅供学习和交流,版权归原文作者所有。如有侵权,请留言联系更正或删除。





相关文章



暂无评论...