







文章导读
做固氮研究时,你大概率默认2-肼基配合物是经典交替路径产物,即1-肼基直接发生近端氮衍生化。几十年来教科书都沿袭这套逻辑,但致命缺陷在于毫无实验支撑,“为何偏偏选中近端氮”的未解之谜让交替路径研究沦为空白。北大团队刚刚推翻了这套共识:2-肼基根本不是近端直接衍生的,而是通过1到2-肼基异构化“变”出来的!这一反直觉机制瞬间打通远端与交替路径壁垒。那个填补缺失一环的肼基阴离子自由基配体,到底藏着怎样跨路径转化的惊人真相?
— 内容由好学术AI分析文章内容生成,仅供参考。
固氮酶的多金属活性位点可以通过缔合路径(通常又分为远端路径和交替路径)实现温和条件下的氮气转化,其中谱学表征和Thorneley-Lowe动力学模型支持了转化过程中存在一种具有二氮烯(diazene)(HN=NH)特征的中间体E4(2N2H),该中间体的两个氮原子各与一个氢原子成键,常被视为典型的交替路径中间体。在合成体系中,人们一直致力于模拟固氮酶的功能以实现温和条件下的氮气转化过程。近几十年来,人们提出了Chatt循环、Schrock循环等经典的远端路径氮气转化机理,而交替路径的研究却非常滞后。具体来说,目前在合成体系中尚不能实现固氮酶中“E4(N2)E4(2N2H)”氮气转化模式,因此文献中从氮气合成二氮烯(或二氮烯的共振结构——2-肼基配合物)的实例非常有限,造成了交替路径机理的研究几乎是空白的。长期以来,人们普遍认为生成二氮烯或2-肼基配合物是通过典型的交替路径实现的——即氮气配合物的两个氮原子交替进行衍生化,其中氮气配合物第一次远端氮原子衍生化的产物(diazenido)直接进行第二次近端氮原子的衍生化生成二氮烯(1-diazene)或2-肼基配合物(2-hydrazido(2-)),但是目前这条路径并没有得到实验证据的支持(图1)。其中的一个未解之谜和关键缺失一环是:为什么第二次衍生化会选择性地发生在近端的氮原子上?因此,研究2-肼基配合物的形成过程对于破译固氮酶的活化机制以及理解何时、为何以及如何从氮气高效获得2-肼基配合物至关重要(图1)。
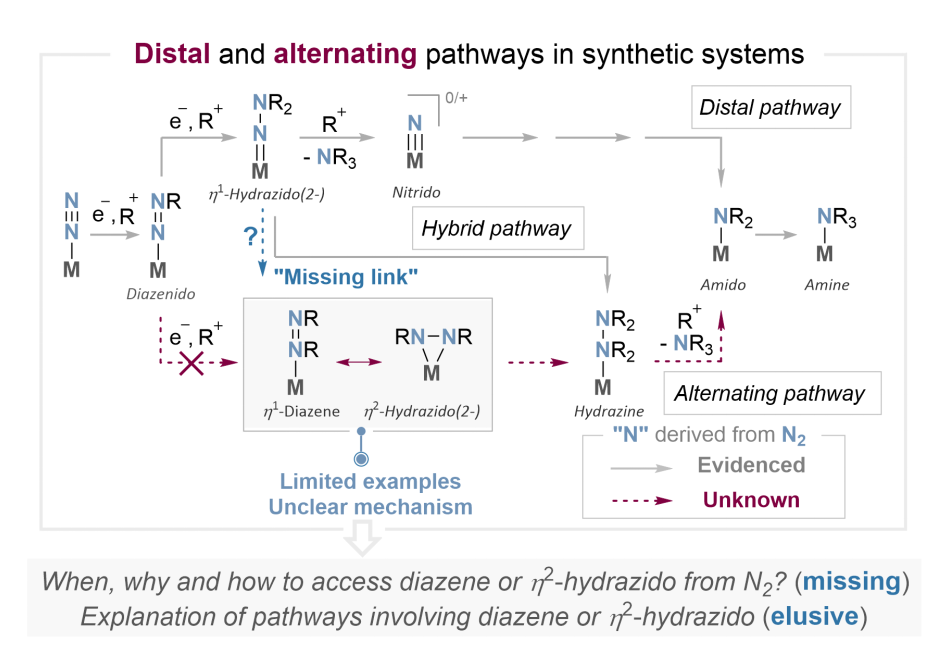
图1 合成体系中的缔合路径
化学与分子工程学院席振峰教授/魏俊年副研究员研究室一直致力于实现温和条件下直接从氮气高效合成含氮有机化合物。近日,围绕金属铬2-肼基配合物的生成是通过二氮烯基配合物直接近端氮原子衍生化(交替路径)还是通过其他未知路径实现的这一关键科学问题,席振峰/魏俊年研究室成功揭示了一种含1/2-肼基异构化的氮气转化新路径,他们明确验证了金属铬2-肼基配合物是通过1-到2-肼基异构化过程生成的,而不是通过经典的交替路径生成的。这一1-到2-肼基异构化过程打通了氮气转化远端路径和交替路径之间的“任督二脉”,不仅回答了2-肼基配合物的生成机理,也填补了氮气转化路径中的关键“缺失一环”。电子结构分析验证了一种肼基阴离子自由基属性的肼基配体,作者以这种关键的电子结构为抓手,合理地设计并从氮气合成了多种类型的2-肼基配合物,为实现多种类型2-肼基配合物的合成和进一步转化提供了条件。该研究核心在于不仅证实了1-/2-肼基异构化反应,更揭示其介导了一种全新的氮气衍生化机制:传统认定的交替路径特征产物(2-肼基配合物),实则可由远端路径中间体(1-肼基配合物)经肼基异构化生成。这种跨路径的转化模式,不仅打破了固氮化学中远端与交替路径的传统壁垒,更提供了一种合成2-肼基配合物的通用策略,实现了从“意外发现”到“理性设计”的跨越。 该工作近期以“A Missing Link in Dinitrogen Fixation Enabled by Hydrazido Isomerization”为题在线发表在J. Am. Chem. Soc.上,文章的第一作者是北京大学博雅博士后殷珠宝,通讯作者是魏俊年,席振峰对该论文进行了指导。该研究得到了国家自然科学基金委、北京分子科学国家研究中心和北京大学化学与分子工程学院的大力支持。
© 版权声明
本文由分享者转载或发布,内容仅供学习和交流,版权归原文作者所有。如有侵权,请留言联系更正或删除。





相关文章



太专业了,这篇完全看不懂,但感觉挺厉害的