







文章导读
你是否想过,科学家能像预测天气一样,精准预测材料的性能?这背后依赖着被称为“第一性原理”的电子结构计算,但它长久以来受困于精度与效率无法兼得的瓶颈。如今,由清华大学徐勇、段文晖研究组领衔的团队,在《自然·计算科学》发表重磅综述,系统揭示了深度学习如何为这一领域带来革命性突破。文章不仅深度剖析了融合AI的高精度量子蒙特卡洛方法,更重点介绍了其代表性的“DeepH”方法——它能将传统计算效率提升数万倍,直指构建“材料大模型”的宏伟目标。这不仅是理论的革新,更是开启数据驱动材料设计与发现新时代的关键钥匙。
— 内容由好学术AI分析文章内容生成,仅供参考。
第一性原理电子结构计算方法是物理、化学、材料科学领域的重要研究手段,但其发展面临精度-效率两难瓶颈。近年来,深度学习技术的迅速发展为该领域提供了新的突破路径。清华大学物理系徐勇、段文晖研究组自2021年年初提出并系统发展“深度学习哈密顿量”(DeepH)方法,成为该方向的代表性方法之一。近日,该研究组与北京大学物理学院陈基研究组合作发表“深度学习电子结构计算”综述文章,推动电子结构计算在复杂量子现象研究以及数据驱动材料发现与设计中的应用。
第一性原理电子结构计算是量子力学的核心挑战问题,提高计算预测的“精度”与“效率”、拓展第一性原理研究的深度和广度是该领域发展的重要目标。量子蒙特卡洛(QMC)方法旨在精确刻画多体强关联效应,而密度泛函理论(DFT)则在单体近似框架下实现对实际材料体系的高效模拟。二者在各自适用范围内广泛应用,分别是“高精度”“高效率”电子结构模拟的代表性计算方法。
本综述重点关注深度学习(DL)与QMC和DFT的交叉前沿,阐述其如何分别在“精度”与“效率”两个维度突破传统第一性原理计算的限制。传统QMC的精度受限于波函数拟设的表达能力,而DL-QMC利用神经网络参数多、表达能力强的特点,进一步提升QMC计算精度。自2017年提出以来,DL-QMC先后推广至周期性体系计算、激发态计算、含时演化等应用场景,在多种体系中精度接近计算化学“金标准”,并成功应用于强关联物态研究。
DL-DFT方法则通过学习原子结构到电子结构基本物理量(如电荷密度、哈密顿量和密度矩阵)的映射关系,跳过传统DFT的自洽求解过程,实现电子结构性质的直接预测。该类方法在网络设计中融合了量子近视性、协变性等物理先验,显著提升了模型的泛化能力,并初步构建出了具备跨元素泛化能力的通用模型。以DeepH为代表的DL-DFT方法将第一性原理电子结构计算拓展至万原子量级,相比传统方法实现了4~5个数量级的加速,并将效率优势延伸至更高精度DFT理论及电声耦合等计算中。
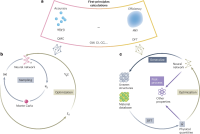
(a)第一性原理电子结构计算以精度与效率为核心目标,其中量子蒙特卡洛(QMC)和密度泛函理论(DFT)分别是代表性的高精度、高效率的计算方法。(b)深度学习QMC原理示意图。(c)深度学习DFT原理示意图
深度学习电子结构方法正经历快速发展。DeepH方法作为DL-DFT领域的代表性方法之一,经历多年更新迭代,正朝着材料领域通用电子结构模型(“材料大模型”)方向发展。这一背景下,DL-DFT与DL-QMC等高精度方法的融合互补,有望弥补DFT在激发态、强关联物态问题方面的不足,共同推动电子结构计算在复杂量子现象研究以及数据驱动材料发现与设计中的应用。
12月22日,该综述以“深度学习电子结构计算”(Deep-learning electronic structure calculations)为题发表于《自然·计算科学》(Nature Computational Science)。
清华大学物理系教授徐勇、段文晖,北京大学物理学院副教授陈基为论文共同通讯作者,清华大学物理系2023级博士生唐泽宸、北京大学物理学院2021级博士生陈浩翔为论文共同第一作者。研究得到国家自然科学基金委卓越研究群体项目、国家科技部重点研发计划、国家自然科学基金重点项目、国家自然科学基金青年学生基础研究项目(博士研究生)、天津超算中心等的支持。
论文链接:
https://www.nature.com/articles/s43588-025-00932-4
供稿:物理系
编辑:李华山
审核:郭玲
© 版权声明
本文由分享者转载或发布,内容仅供学习和交流,版权归原文作者所有。如有侵权,请留言联系更正或删除。







相关文章



从2017年发展到现在的通用模型,进展真快,给研究组点个赞!
希望这些方法能快点应用到新材料发现上,解决一些卡脖子问题。
这综述发在《自然·计算科学》上,水平肯定很高,国内科研越来越牛了。
看起来好专业,我这个外行有点看懵了,有更通俗的解释吗?🤔
DeepH方法能加速这么多,以后材料研究效率会大大提升吧。
深度学习在物理计算中的应用越来越广了,期待看到更多实际成果。
清华和北大的合作真是强强联合啊!👍