






文章导读
当大脑遭遇癫痫或脑损伤,锌离子(Zn2+)竟会“反客为主”,触发神经元异常兴奋?西安交大王昌河团队与川大赖颖团队联合发现,病理状态下升高的Zn2+并非旁观者,而是直接结合突触融合关键蛋白Syt1的C2A-C2B界面,以纳摩尔级亲和力改变其构象,促进突触囊泡过度对接,导致神经递质自发释放失控。这一机制揭示了Zn2+致神经毒性的关键路径,为神经系统疾病的治疗提供了全新靶点。
— 内容由好学术AI分析文章内容生成,仅供参考。
在中枢神经系统中,神经元的结构与功能完整性是维持正常认知与行为活动的基础。然而,多种病理状态(如癫痫、短暂性脑缺血、脑损伤等)均可能导致神经元结构破坏与功能障碍。在这些病理条件下,神经元内源性锌离子(Zn2+)的浓度显著升高,从而引发一系列神经毒性效应。已有研究表明,Zn2+在神经元内的异常积累是介导神经毒性的重要因素,其可能通过调控细胞膜通透性、破坏离子稳态、干扰离子通道功能、影响神经递质释放以及破坏信号传导通路等多种机制,造成神经元的功能紊乱。尽管Zn2+在神经损伤中的作用受到广泛关注,其确切的分子机制仍未完全阐明。因此,本研究旨在在特定病理状态下,系统探讨Zn2+在调控神经元信号传导过程中的作用机制,以期为理解其在神经病理状态中的致病作用提供新的理论依据。
8月2日,西安交大生命学院王昌河教授团队联合四川大学赖颖研究员团队在《自然通讯》(Nature communications)期刊上发表了最新研究成果《突触结合蛋白-1作为主要的锌离子传感器,在病理条件下介导神经递质的自发释放》(Synaptotagmin-1 serves as a primary Zn2+ sensor to mediate spontaneous neurotransmitter release under pathological conditions)。研究发现,当神经元内Zn2+浓度升高时,可显著增强神经递质的自发释放(spontaneous neurotransmitter release)。进一步的机制研究发现,突触融合关键调控蛋白Synaptotagmin 1(Syt1)能够与Zn2+发生相互作用,通过影响突触囊泡的融合与释放,从而改变突触传递效率与神经元间的信号通信。这一发现不仅揭示了Zn2+在神经元信号调控中的潜在作用机制,也为进一步研究Zn2+在神经系统疾病中的致病机制提供了重要的理论依据和研究起点。

Zn2+促进海马神经元中的微小兴奋性突触后电流(mEPSC)
研究发现,向体外培养海马神经元的细胞外液中注入Zn2+,可观察到其以剂量依赖的方式显著增加微小兴奋性突触后电流(mEPSC)的频率,但mEPSC的幅值未发生变化,提示Zn2+在静息状态下主要通过增强突触前囊泡的释放概率发挥作用。相比之下,相比之下,Zn2+对诱发性兴奋性突触后电流(evoked EPSC)幅度未产生显著影响,进一步支持其作用机制偏向于调控自发释放事件的频率而非单一突触事件的强度。此外,当在海马神经元中敲除Synaptotagmin 1(Syt1)后,发现Zn2+对自发释放的增强作用被显著抑制,表明Syt1在Zn2+介导的突触释放调控中起关键作用。这些结果提示,Zn2+可能通过与Syt1相互作用,选择性调节突触小泡的释放频率,从而参与调控突触前活动和神经元之间的信息传递。
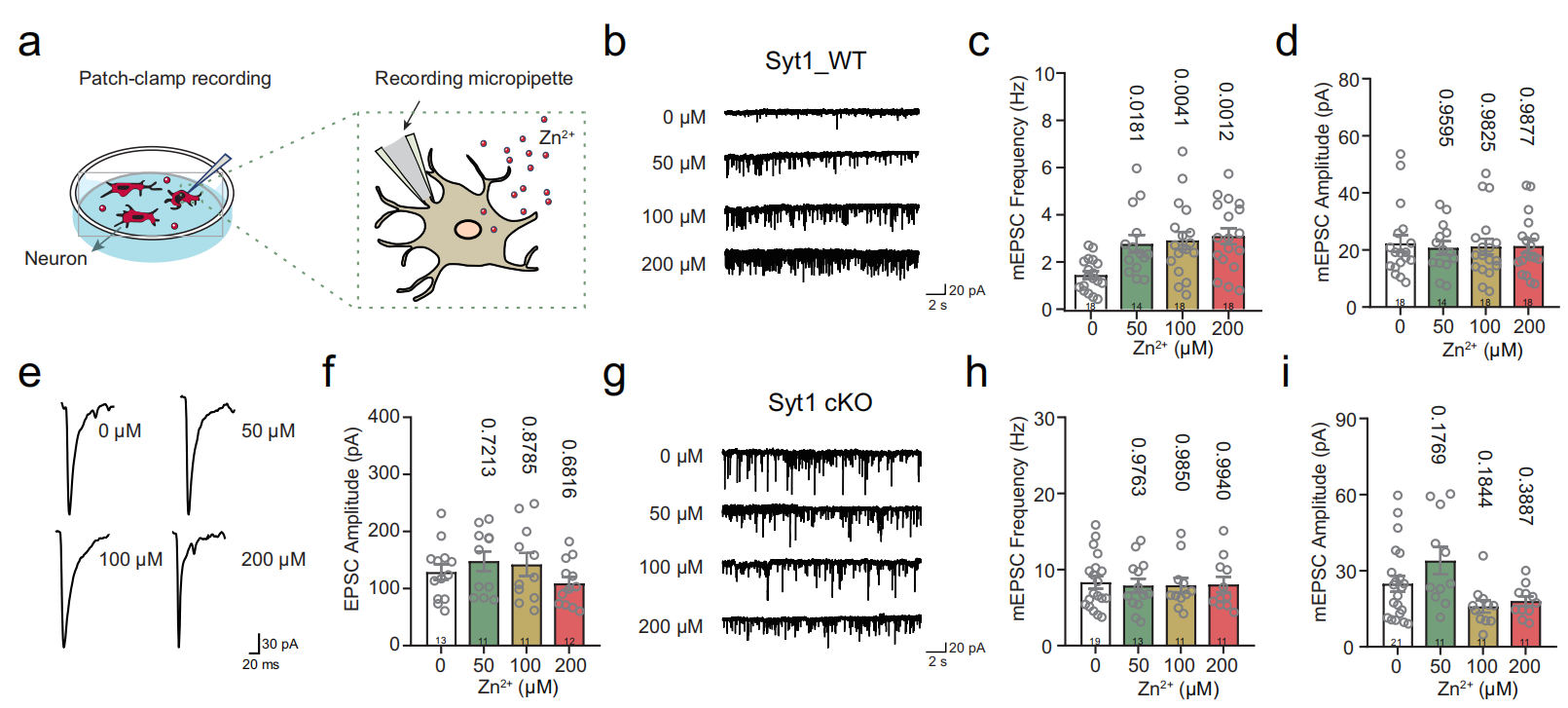
图1:Zn2+促进海马神经元的自发分泌
Zn2+与C2AB结合,Zn2+与C2AB的结合方式与Ca2+存在显著差异
研究通过Zn2+分别结合C2AB/C2A/C2B引起Zn2+指示剂改变溶液中荧光强度的变化,观察Zn2+与C2AB的结合。其结果显示Zn2+与C2A和C2B之间的界面结合。此外,通过比对Syt1与Syt3的基因序列预测C2AB与Zn2+的结合位点,结合荧光指示剂实验验证其结合位点为D261/E386/H389。还通过突变C2AB的Ca2+结合位点,表明Zn2+与C2AB结合方式与Ca2+不同。
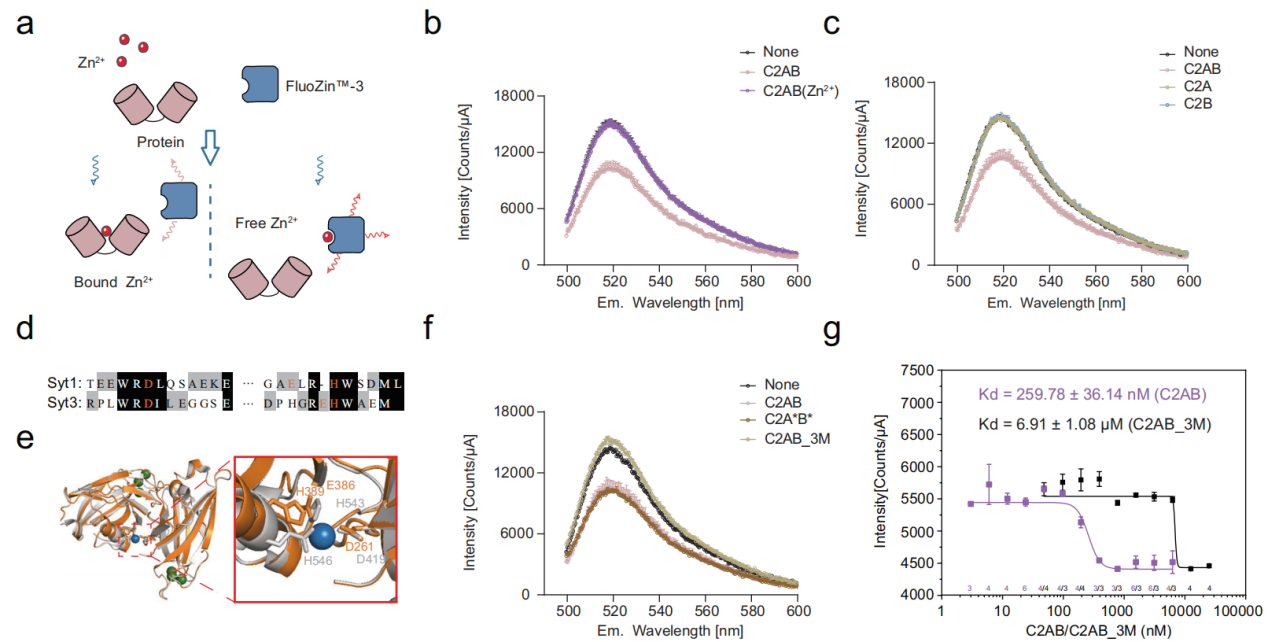
图2:锌离子与C2A-C2B结构域的界面结合,结合亲和力为纳摩尔(nM)级
Zn2+结合Syt1增强突触小泡囊泡的对接
研究团队进一步研究了Zn2+与Syt1结合增强神经元兴奋的机制。通过在海马细胞中表达synaptophysin-pHluorin(Syp-pH),并利用实时全内反射荧光显微镜(TIRF)成像技术监测突触末梢中突触小泡的动态对接过程。实验中,当将Zn2+灌注至细胞外液后,荧光信号在突触前末梢显著增强,提示囊泡数量向质膜靠近并稳定对接的事件显著增加。该过程与Zn2+转移至突触前区域同时发生,表明Zn2+促进突触小泡对接,从而提高神经递质释放效率。进一步结合Syt1敲除实验结果支持,Zn2+可能通过与Syt1相互作用,增强突触小泡对接过程,从而在突触前水平促进突触传递的增强。。
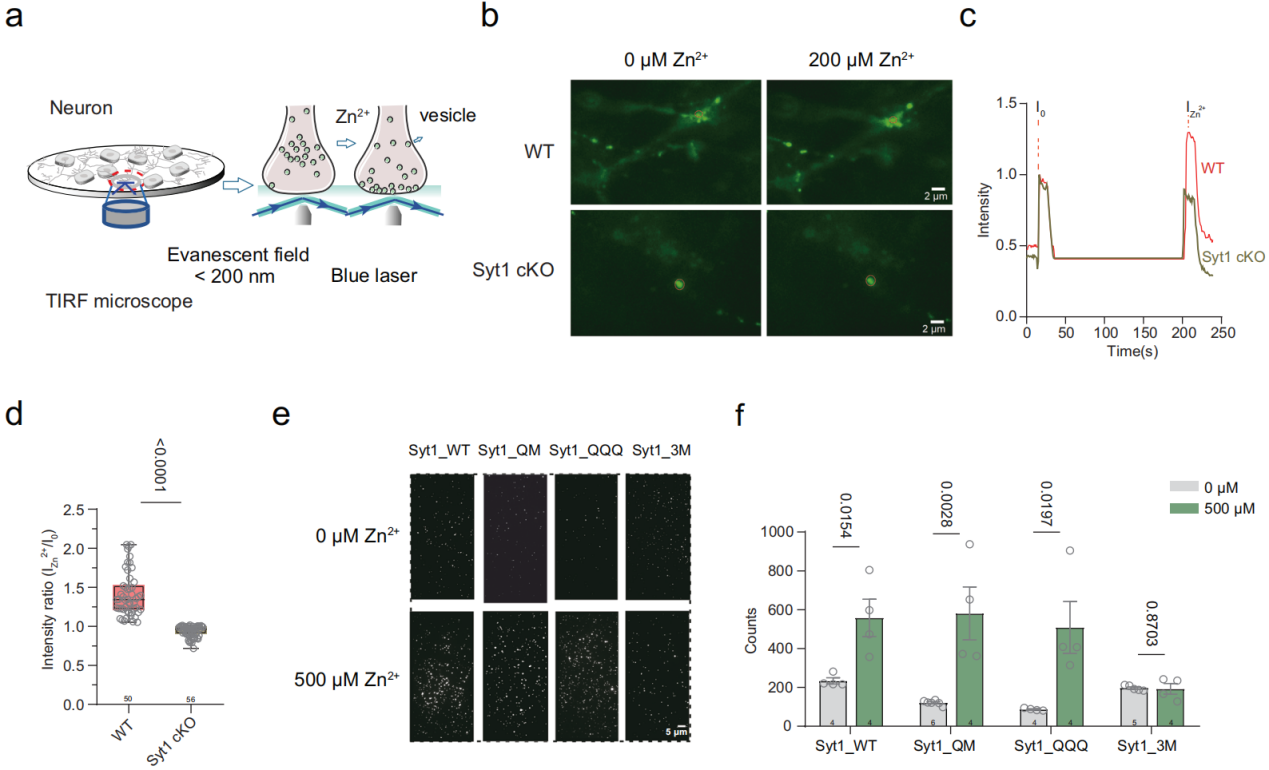
图3:锌离子与Syt1结合促进囊泡的对接
该研究最终表明,Zn2+在病理条件下对Synaptotagmin 1(Syt1)的调控机制与其在生理状态下参与自发释放的模式存在显著差异。在生理条件下,自发释放被抑制在基线水平,直至胞内Ca2+升高并结合Syt1,促使其与SNARE复合物协同介导突触小泡融合。而在病理状态下,细胞内Zn2+浓度升高时,Zn2+可能结合到Syt1的C2A-C2B结构域界面结合,改变其构象并增强了其对阴离子膜的结合能力,促进囊泡与突触前膜的对接,从而诱导突触前自发释放事件异常增加。
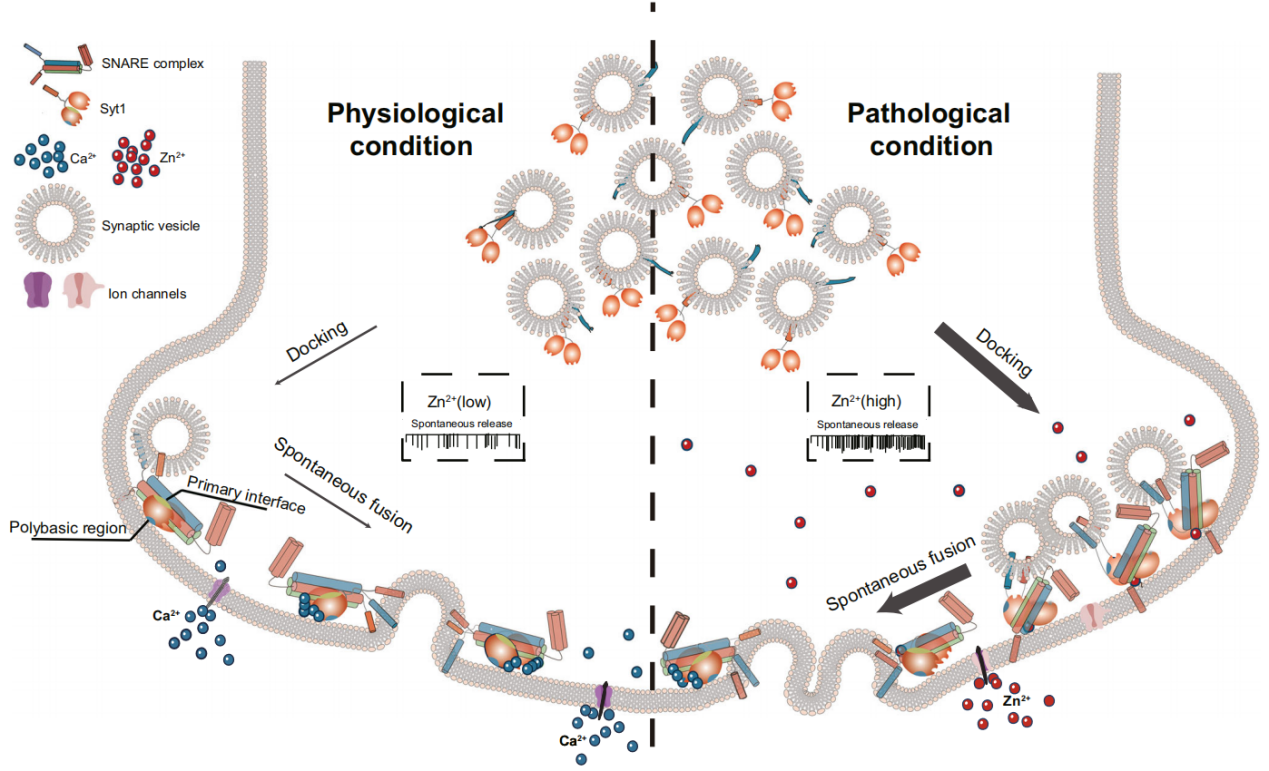
图4:在生理/病理条件下Zn2+-Syt1互作调控神经递质自发释放过程
本研究首次提出,Syt1-Zn2+复合物的构象在Syt1介导的病理性自发神经递质释放过程中具有关键调控作用。该研究发现了一个简洁而新颖的Syt1-Zn2+相互作用模型,揭示了在癫痫发作、短暂性脑缺血或创伤等病理条件下,内源性Zn2+浓度升高如何通过作用于Syt1,促进突触小泡对接与异常释放,进而导致神经元的过度兴奋与毒性反应。该研究为深入理解Zn2+在神经系统疾病中的作用提供了重要的理论基础,也为相关疾病的干预策略提供了潜在靶点。
四川大学向易娟博士、崔乐乐博士、西安交大姚靖宇博士和爱荷华州立大学娄晓初博士为本文的共同第一作者,四川大学赖颖研究员和西安交大王昌河教授为本文的共同通讯作者。本研究得到国家自然科学基金、陕西省重点研发计划、西安交大重大项目等项目的支持。
原文链接: https://www.nature.com/articles/s41467-025-62496-1
© 版权声明
本文由分享者转载或发布,内容仅供学习和交流,版权归原文作者所有。如有侵权,请留言联系更正或删除。





相关文章




暂无评论...