
研究实现未保护酚类化合物高效三氟甲基化







文章导读
你在合成药物时,总是被酚/胺的保护‑脱保护步骤拖慢进度,常规金属催化又难以控制三氟甲基的邻位位点。青岛团队借助双功能小分子协同改造的P450酶,让未保护酚或芳胺直接定位生成C‑CF₃键,选择性高达99%,底物转化数突破6700,几乎不受官能团影响。实验已在半放大尺度合成关键药物中间体,展示了从有机氟化到生物催化的全新桥梁。这种方法能为你的项目省去保护步骤、提升产率,你想先了解它的核心协同机制吗?
— 内容由好学术AI分析文章内容生成,仅供参考。
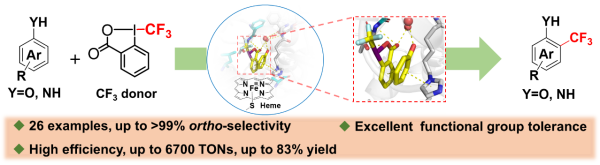
三氟甲基作为药物化学中重要的修饰基团,引入该基团可显著提升候选分子的代谢稳定性和生物活性。在未保护的酚和苯胺类化合物上直接实现高区域选择性三氟甲基化,是合成化学领域长期存在的技术难题。传统化学方法常依赖导向基团或金属催化剂,反应条件苛刻,选择性欠佳。
中国科学院青岛生物能源与过程研究所研究团队基于前期构建的“双功能小分子协同P450酶催化”体系,通过双功能小分子与蛋白质工程改造的协同调控,精准预组装酚/芳胺底物与三氟甲基化试剂的结合构象,严格限定C-CF3键的形成位点。该策略突破了自由基偶联反应区域选择性不足的限制,首次实现了未保护酚和芳胺类化合物的高邻位区域选择性三氟甲基化。实验数据显示,反应选择性最高可达99%,底物催化转化数突破6700。
该生物催化体系展现出优异的底物普适性和官能团耐受性,可适用于不同取代的苯酚、苯胺及杂芳香族酚/胺底物。分子动力学模拟揭示,酶突变体与双功能小分子通过协同调控底物构象,进而控制区域选择性的分子机制。半制备规模实验已成功制备出2-三氟甲基-4-氰基苯酚和3-羟基-2-三氟甲基吡啶两种关键药物中间体,验证了该体系的合成应用潜力。
该研究构建了生物催化与有机氟化学的桥梁,建立了一种含氟生物活性分子合成的新范式,为药物研发和功能材料创制提供了新工具。
相关研究成果发表在《德国应用化学》(Angewandte Chemie International Edition)上。研究工作得到了国家重点研发计划、国家自然科学基金、山东省泰山学者计划等的支持。
研究实现未保护酚类化合物高效三氟甲基化
© 版权声明
本文由分享者转载或发布,内容仅供学习和交流,版权归原文作者所有。如有侵权,请留言联系更正或删除。





相关文章




感觉离实际应用还远,不过思路挺新颖的
底物普适性怎么样?苯环上带强吸电子基能行不?
又是P450酶,这玩意儿真是万能工具啊
看不懂但大受震撼,给科研人员点赞👍
所以这个双功能小分子是啥?能具体说说吗🤔
之前搞过类似反应,条件苛刻得要命,这个酶催化确实牛
选择性99%?这数据有点猛啊,不知道能不能工业化
这啥啊,化学名词一堆,但感觉好厉害的样子😂