
科研人员研发出新型嘌呤类PGK1抑制剂







文章导读
你是否知道,全球数百万炎症性肠病患者正面临现有药物副作用的困扰?中科院团队最新突破或许将改变这一现状。研究人员通过精准药物设计,成功开发出新型嘌呤类PGK1抑制剂——化合物6e,该分子不仅能高效阻断导致肠道炎症的关键代谢通路,更在动物实验中展现出惊人的治疗效果:口服即可显著缓解结肠炎症状,且具有优异的选择性和生物利用度。这项发表于国际权威期刊的研究,为开发更安全有效的炎症性肠病靶向药物开辟了全新路径。
— 内容由好学术AI分析文章内容生成,仅供参考。
近日,中国科学院合肥物质科学研究院健康与医学技术研究所刘青松团队,研发出新型嘌呤类磷酸甘油酸激酶1(PGK1)抑制剂。这一化合物在炎症性肠病模型中表现出良好的治疗效果。
炎症性肠病是慢性、复发性肠道炎症疾病。现有治疗药物均伴有副作用。近年来,PGK1作为糖酵解过程中的关键代谢酶,被发现是炎症性肠病的潜在治疗靶点。然而,现有PGK1抑制剂在活性或药代动力学性质方面的局限性,限制了其临床应用。因此,发展具有新型骨架及良好成药性的PGK1抑制剂具有重要意义。
研究团队在前期通过高通量筛选发现的苗头化合物NG52的基础上,采用基于结构的理性药物设计,对嘌呤衍生物NG52的R1、R2和R3区域开展构效关系研究,合成了一系列新化合物,并检测它们对PGK1的抑制活性。
团队经过多轮结构优化发现,嘌呤衍生物6e在酶活水平上表现出优异活性,并在大鼠和小鼠中均展现出良好的生物利用度和较高的口服暴露量。同时,在激酶选择性评估中,6e在210种激酶中对PGK1具有高选择性。体外研究显示,6e抑制PGK1介导的糖酵解过程,降低葡萄糖消耗和乳酸生成,上调Nrf2蛋白,促进HO-1蛋白表达,抑制IL-1β和IL-6的转录与蛋白水平,从而发挥抗炎作用。在动物结肠炎模型中,口服6e能够剂量依赖性地缓解体重下降、降低疾病活动指数、改善结肠缩短和组织病理损伤,表现出良好的体内抗炎疗效。
这一研究为PGK1靶向治疗炎症性肠病提供了具有潜力的临床前候选药物。
相关研究成果在线发表在《药物化学杂志》(Journal of Medicinal Chemistry)上。研究工作得到国家自然科学基金等的支持。
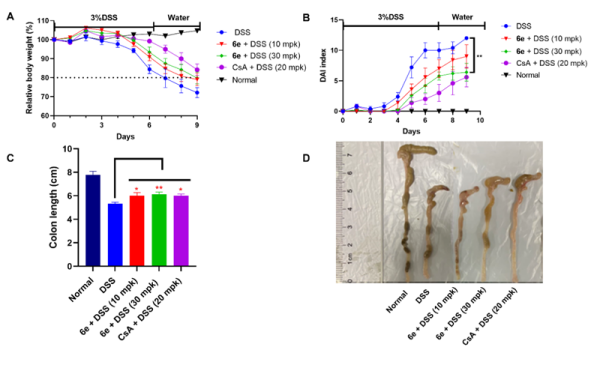
化合物6e在DSS诱导的小鼠急性结肠炎模型中的治疗效果
© 版权声明
本文由分享者转载或发布,内容仅供学习和交流,版权归原文作者所有。如有侵权,请留言联系更正或删除。





相关文章





暂无评论...