







文章导读
在有机合成中,如何精准控制复杂分子中官能团的位点?传统方法受限于双键位置,多取代烯烃的位点选择成为长期难题。武汉大学阴国印课题组在Nature Chemistry发表突破性研究,揭秘一种新型链识别策略:借鉴生物识别概念,设计空间位阻梯度配体,实现了多取代烯烃头尾碳硼化反应的高位点选择性精准调控。这一创新基于金属迁移催化技术,颠覆了传统双官能化的局限,可高效合成天然产物和有机太阳能电池关键中间体,为药物开发与功能材料领域开辟革命性新路径。阅读全文,了解这一策略如何破解复杂分子构筑的瓶颈。
— 内容由好学术AI分析文章内容生成,仅供参考。
(通讯员高妍)近日,武汉大学高等研究院、复杂生命体代谢与调控全国重点实验室与泰康生命医学中心教授阴国印课题组在国际期刊Nature Chemistry上在线发表了题为“Head–tail carboboration of multisubstituted alkenes enabled by chain recognition”的研究论文。该研究发展了一种新型链识别策略,实现了对多取代烯烃头尾位点选择性碳硼化反应的精准控制。高等研究院2022级博士生孔维宇、2020级博士生吴冬和2019级硕士生魏红为论文共同第一作者,阴国印与副研究员李阳阳为共同通讯作者。武汉大学高等研究院为论文第一完成单位。
在有机合成中,将官能团精确引入碳链的预设位置,一直是化学家追求的核心目标。然而,对于多取代烯烃这类结构复杂的分子,由于存在多条相似碳链以及多个反应性相近的潜在位点,要实现位点选择性的精准调控尤为困难。传统烯烃双官能化策略虽然高效,但其反应模式通常受限于双键的固有位置,灵活性和普适性有限。
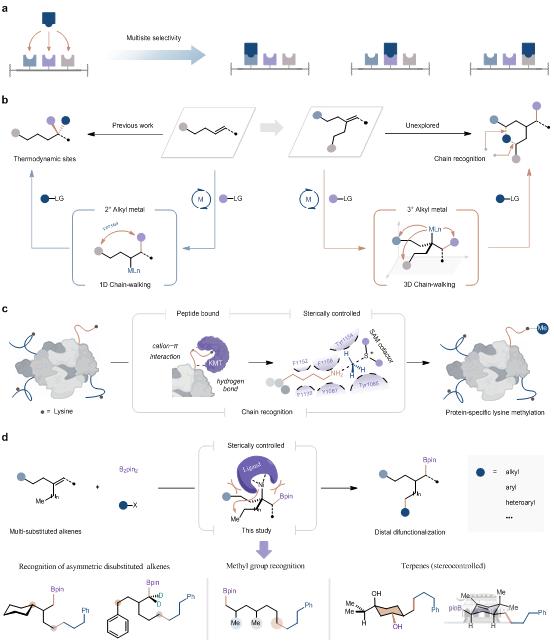
多取代烯烃位点选择的挑战、生物链识别的启发,以及本研究提出的空间受控链识别模型
阴国印课题组基于其先前发展的金属迁移催化策略(Science2022,376,749-753;Acc. Chem. Res.2023,56,3246-3259),借鉴生物识别中的链识别概念,通过设计具有空间位阻梯度的配体,成功突破了传统双官能化反应对双键位置的依赖,实现了多取代烯烃的精准位点选择性调控。在此基础上,他们开发了多取代烯烃的高位点选择性头尾碳硼化反应。该方法不仅实现了多种碳亲电试剂与复杂烯烃底物的高效偶联,还可应用于天然产物和功能分子(如有机太阳能电池关键中间体)的简洁合成,为复杂分子的精准构筑提供了新思路,有望在药物合成和功能材料等领域展现出广阔的应用前景。
该研究得到国家自然科学基金、中央高校基本科研业务费专项资金、青年教师科研创新能力培育项目以及广东省基础与应用基础研究基金等资助。高等研究院闵杰教授课题组在有机太阳能电池关键中间体合成方面提供支持。测试工作由武汉大学科研公共服务条件平台完成,DFT计算获武汉大学超算中心支持。
论文链接:https://www.nature.com/articles/s41557-025-01903-y
(供图:高等研究院 编辑:相茹)
© 版权声明
本文由分享者转载或发布,内容仅供学习和交流,版权归原文作者所有。如有侵权,请留言联系更正或删除。





相关文章



暂无评论...