







文章导读
PROTAC技术虽已进入临床,但E3连接酶配体匮乏、VHL抑制可能带来风险等问题长期制约其发展。上海交通大学沈玉道与赵博课题组基于VHL与RIPK1形成内源性复合物的发现,另辟蹊径提出RIMTAC平台,通过RIPK1抑制剂间接招募VHL,规避了传统VHL配体药代动力学差和抑癌功能丧失的缺陷。他们设计出靶向BRD4、AKT、JAK1的降解剂,其中化合物10对BRD4的DC50仅61.75 nM,最大降解率达80%,且降解严格依赖泛素-蛋白酶体系统和四元复合物形成。这种融合RIPK1抗炎活性的新策略,能否为蛋白降解工具拓展和炎症性疾病治疗开辟全新路径?
— 内容由好学术AI分析文章内容生成,仅供参考。
PROTAC(蛋白降解靶向嵌合体)技术为创新药物研发开辟了全新路径。截至目前,全球已有超过20种PROTAC分子进入临床研究,覆盖多种实体瘤和血液系统恶性肿瘤,其中ARV471已经获批上市。相较于传统小分子抑制剂,PROTAC具备高选择性、低剂量催化降解、可克服获得性耐药以及靶向“不可成药”蛋白等显著优势,展现出广阔的治疗前景。然而,PROTAC的广泛应用长期受限于可用的E3泛素连接酶配体种类稀少。在人类已鉴定的600余种E3连接酶中,目前仅少数可通过小分子配体进行有效招募。其中,VHL是与CRBN并列的最常用于PROTAC设计的E3连接酶,但现有VHL配体(抑制剂)普遍存在药代动力学性质不佳的问题,且VHL本身作为肿瘤抑制因子,被抑制可能带来潜在的生物学风险。因此,开发新型VHL配体或探索间接招募VHL的策略,对于拓展PROTAC工具箱、提升其成药性和安全性具有重要意义。

沈玉道课题组和赵博课题组合作提出并验证了一种新型靶向蛋白降解平台—RIMTAC(RIPK1-Mediated TArgeting Chimera),该平台通过间接招募VHL E3连接酶实现靶向蛋白降解,为PROTAC技术领域提供了重要拓展。研究团队基于VHL与RIPK1在内源性条件下形成复合物的发现,创新性地采用RIPK1抑制剂作为VHL的间接招募工具。通过生物素下拉实验证实,RIPK1抑制剂与RIPK1的结合并不干扰RIPK1与VHL的相互作用,为该策略提供了关键理论支撑。
作为概念验证,研究者设计了靶向BRD4、AKT和JAK1的系列RIMTAC分子,经系统筛选获得了代表性化合物10(BRD4降解剂)、14(AKT降解剂)和24(JAK1降解剂)(图1)。这些化合物均能诱导浓度和时间依赖性的靶蛋白降解,其中化合物10对BRD4的DC50为61.75 nM,最大降解率达80%(图2)。全面的机制研究表明,RIMTAC介导的降解严格依赖泛素-蛋白酶体系统(MG132和MLN4924可完全逆转降解效应),且降解过程不改变靶蛋白mRNA水平(图3)。更重要的是,竞争实验、敲低实验及免疫共沉淀结果一致证明,功能性降解需要BRD4、RIMTAC分子、RIPK1与VHL形成四元复合物(图4)。
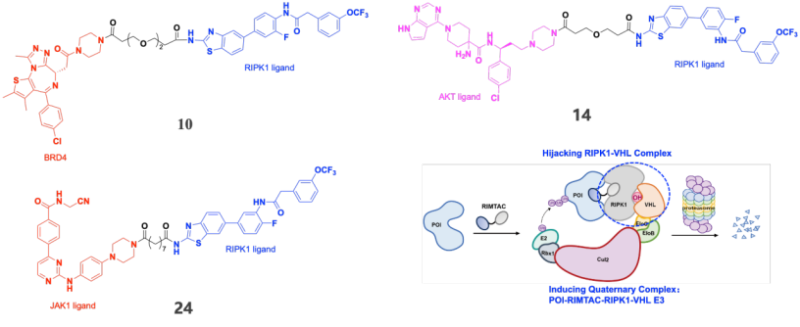
图1. BRD4、AKT、JAK1 的RIMTAC降解剂结构及RIMTAC示意图
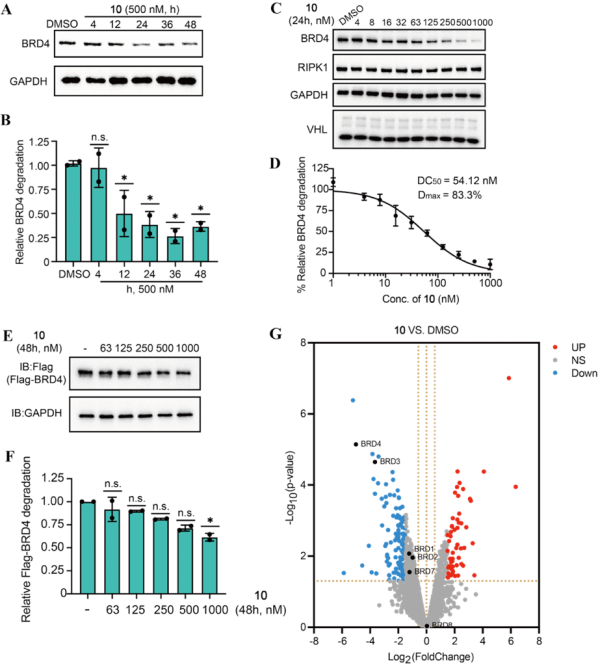
图2. BRD4降解活性和选择性表征
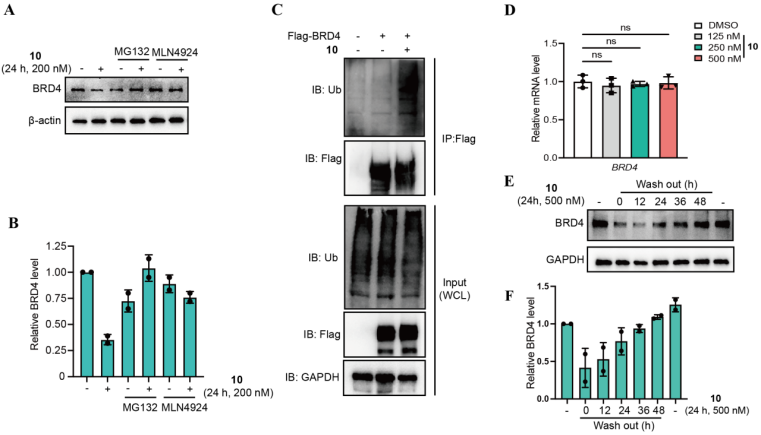
图3. BRD4降解依赖于泛素-蛋白酶体系统
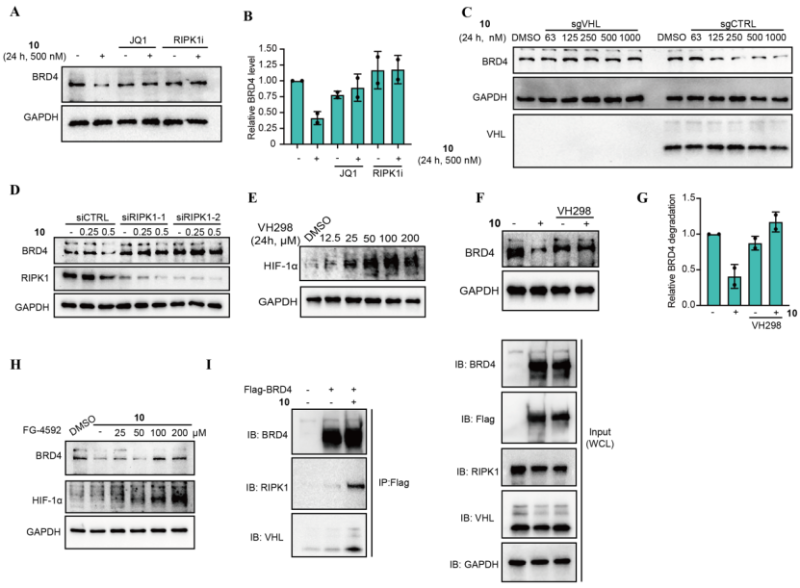
图4. BRD4降解依赖于BRD4-10-RIPK1-VHL形成的四元复合物
RIMTAC平台成功实现了通过靶向E3连接酶结合蛋白来间接招募E3连接酶的降解策略,有效规避了传统VHL配体口服生物利用度低的成药性缺陷和保留了VHL作为肿瘤抑制因子的功能。同时,RIPK1抑制剂本身具有抗炎活性,与靶向降解BRD4、JAK1等炎症介质的效应相结合,展现出潜在独特的协同治疗潜力。该研究不仅拓展了靶向蛋白降解的工具箱,也为炎症性疾病治疗提供了新思路。
该工作得到了国家自然科学基金、科技部重点研发计划、上海市药物靶标发现及递送前沿科学研究基地等基金的支持。上海交通大学药学院沈畅和孙汉寅两位博士研究生为本论文的共同第一作者,沈玉道长聘教轨副教授、朱明彦讲师和赵博长聘副教授为本论文的共同通讯作者。
原文链接:https://pubs.acs.org/doi/10.1021/acs.jmedchem.6c00897
作者: 药学院 供稿单位: 药学院
© 版权声明
本文由分享者转载或发布,内容仅供学习和交流,版权归原文作者所有。如有侵权,请留言联系更正或删除。





相关文章


这个RIMTAC思路挺巧,避开传统配体缺陷