







文章导读
你正为光催化把二氧化碳只产出CO、CH4等单碳产品而抓狂吗?大多数催化剂因为缺乏缺陷位和有效的C‑C偶联路径,选择性始终低于30%。南京大学团队利用非晶FeOx介孔纳米片,通过提升氧空位与Fe²⁺/Fe³⁺比例,实现光照520 nm下表观量子效率1.60%,乙烷选择性最高达94%。这套结构‑电子协同调控或许正是突破多碳燃料瓶颈的钥匙——你想先了解它背后的“隐藏机制”,还是等到别人抢先发布?
— 内容由好学术AI分析文章内容生成,仅供参考。
南京大学物理学院环境材料与再生能源研究中心邹志刚院士和周勇教授课题组与东南大学合作,设计合成了独特的非晶FeOx介孔纳米片光催化剂,通过结构-电子协同调控实现了CO2高选择性光催化转化为乙烷(C2H6)。该催化剂在520 nm处表观量子效率达1.60%,乙烷选择性达约70%(产率基准)和约94%(电子基准)。
将CO2转化为高能量密度烃类燃料是实现碳循环与碳中和的重要途径之一。然而,由于C=O键的高稳定性,目前光催化CO2还原系统主要生成C1产物(如CO、CH4、CH3OH),而高效、高选择性地生成高附加值的多碳(C2+)烃类仍面临诸多挑战,包括充足的电子-空穴对、稳定的反应中间体、适宜的C-C偶联能垒以及高效的电荷分离与传输。
针对上述难题,研究团队提出了结构-电子协同调控的设计策略。非晶材料因其缺乏长程原子有序性,具有比晶态材料更高的缺陷密度和不饱和配位位点,可通过调控原子结构来优化电子性质和化学环境。同时,介孔材料具有丰富的相互连通孔道,可显著增加比表面积,为吸附分子提供更多可接触活性位点。FeOx因其地壳丰度高、化学稳定性好、铁氧化态可调(Fe2+/Fe3+)以及缺陷化学可调控等特点,成为整合上述结构优势的理想平台。
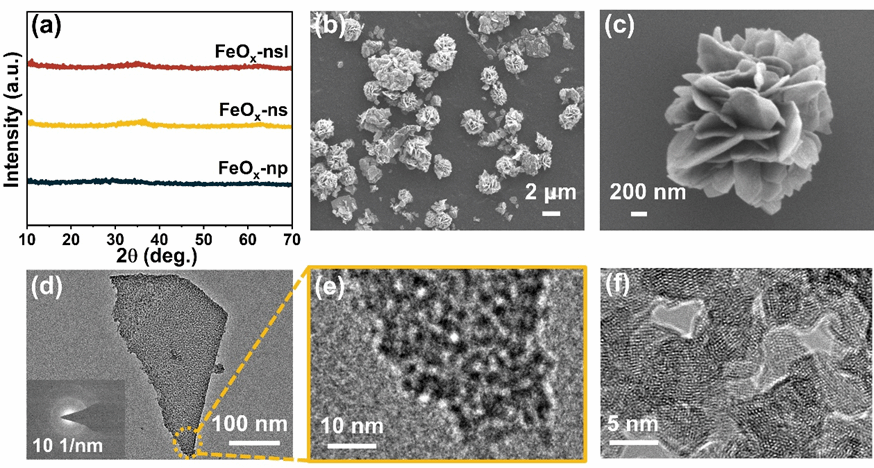
图1 (a) FeOx 的 XRD 图谱。FeOx-ns 的 (b-c) SEM,(d-f) TEM 图像(插图:SAED 图案),HRTEM。
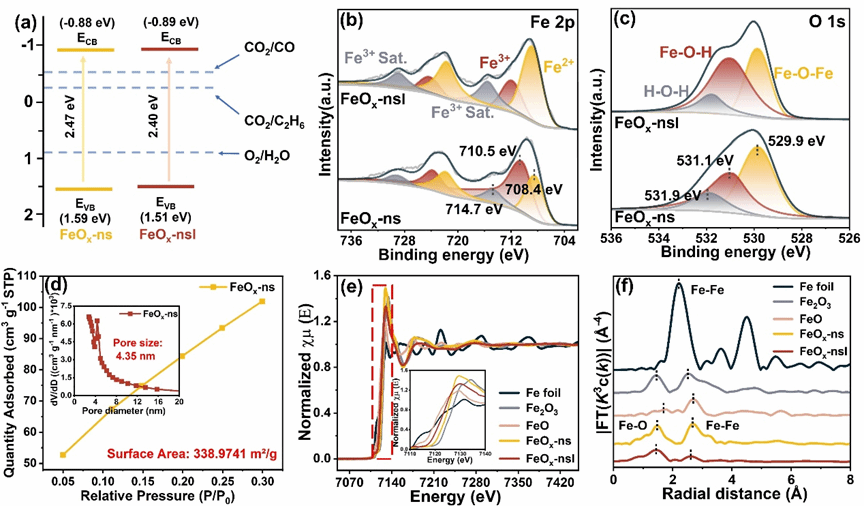
图2 (a) 能带位置示意图。样品的 (b) Fe 2p 和 (c) O 1s XPS 光谱。(d) FeOx-ns 的氮吸附等温线(插图:孔径分布)(e) Fe K 边 XANES 光谱(插图:红色方框的放大图)以及 (f) 样品 Fe K 边 EXAFS 光谱的傅里叶变换。
本研究成功制备了非晶FeOx介孔纳米片(FeOx-ns),并通过脉冲激光刻蚀处理得到FeOx-nsl,在保持非晶特征的同时增加了氧空位浓度和Fe2+/Fe3+比例。该催化剂呈现花状层级微结构,由厚度约20-40 nm、平面尺寸约800 nm-2 μm的纳米片组成,纳米片内含有平均孔径3-4 nm、壁厚4-5 nm的均匀介孔。BET测试显示其比表面积达339 m2/g,CO2吸附量为24.42 cm3/g。
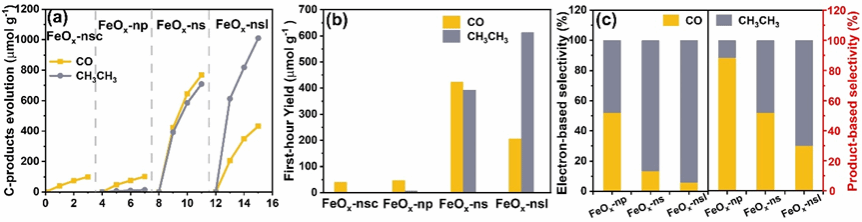
图3 (a) C产物的演变,(b) FeOx-nsc、FeOx-np、FeOx-ns和FeOx-nsl在第一小时的光催化活性比较。(c) FeOx-np、FeOx-ns和FeOx-nsl的电子选择性和产物选择性。
光催化性能测试表明,FeOx-nsl在第一小时内乙烷产率高达612.69 μmol g-1,而CO产率降至206.01 μmol g-1。产率和电子基准的乙烷选择性分别达到约70%和约94%,表观量子效率在520 nm处达1.60%,是FeOx-ns(0.43%)的近4倍。同位素标记实验(13CO2)证实产物碳源完全来自输入的CO2。
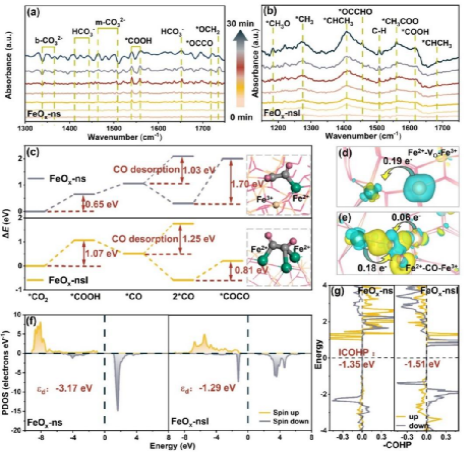
图4 (a) FeOx-ns,(b) FeOx-nsl的原位红外光谱。(c) 针对FeOx-ns和FeOx-nsl的CO2还原生成*COCO的反应能变化,及相应的原子结构配置。(d) Fe2+-Vo-Fe3+和 (e) Fe2+-CO-Fe3+结构的电荷密度差异(黄色表示电子积累,蓝色表示电子耗损)。(f) FeOx-ns和FeOx-nsl的PDOS及d带中心。(g) FeOx-ns和FeOx-nsl的ICOHP。
机理研究结合原位红外表征与密度泛函理论计算(DFT),深入揭示了结构-电子协同调控促进C-C偶联的微观机制。DFT计算表明,CO2在Fe3+位点的吸附能于Fe2+位点,且*COOH在Fe3+位点形成的反应能著低于Fe2+位点,说明Fe3+是CO2活化的主要活性位点。然而,生成的*CO中间体更倾向于吸附在Fe2+位点,且Fe2+位点上C-C偶联形成*COCO的反应能低于Fe3+位点,这为CO从Fe3+向Fe2+迁移并发生C-C偶联提供了热力学驱动力。
氧空位在串联催化中发挥关键作用。电荷密度差分分析显示,Fe2+-Vo- Fe3+构型中氧空位破坏了局部结构对称性,导致0.19个电子从Fe3+向Fe2+转移,形成极化电子结构,促进CO迁移。NEB计算表明该迁移过程反应能为-0.69 eV、动力学能垒仅1.14 eV,热力学和动力学均可行。COHP分析显示Fe2+位点上Fe-(C-O)键的ICOHP值为-1.51 eV,成键更强,有利于稳定CO并促进C-C偶联。原位DRIFTS光谱在FeOx-nsl表面检测到*CH3O、*OCCHO等C2产物特征中间体,与DFT结果相互印证,共同揭示了Fe3+主导CO2活化、Fe2+促进C-C偶联的串联催化机制。介孔结构的空间限域效应进一步富集局部*CO浓度,协同提升了C2产物选择性。
该工作为通过跨尺度结构协同调控实现CO2选择性转化为高附加值多碳燃料提供了设计原理,展示了非晶结构、纳米级孔隙率和可调控氧化还原化学的集体协同效应在光催化CO2还原中的应用潜力。研究成果近期以”Design of Unique Amorphous FeOx Mesoporous Nanosheets for Structural-Electronic Synergy toward Highly Selective and Efficient CO2 Photoconversion into Multicarbon Fuels”为题发表在Journal of the American Chemical Society,(https://doi.org/10.1021/jacs.6c01273)。
该工作由南京大学周勇教授、东南大学李强教授、王金兰教授合作完成,邹志刚院士给与了指导性的建议。南京大学为第一通讯单位。南京大学物理学院博士生成婷婷与东南大学物理学院博士生林旺强为共同第一作者。研究工作得到南京大学物理学院、固体微结构物理全国重点实验室、人工微结构科学与技术协同创新中心、江苏省纳米技术重点实验室和南京大学环境材料与再生能源研究中心等平台支持,获得国家重点研发计划项目、国家自然科学基金、江苏省自然科学基金项目项目等资助。
论文链接:https://doi.org/10.1021/jacs.6c01273
© 版权声明
本文由分享者转载或发布,内容仅供学习和交流,版权归原文作者所有。如有侵权,请留言联系更正或删除。







相关文章





南大这波操作太秀了,乙烷选择性居然能到94%?