科研人员揭示患者源性EGFR R252C突变促进肿瘤进展新机制







文章导读
你正为家人携带EGFR罕见突变却无药可治而彻夜难眠,看着标准靶向药一次次失效,误以为这类突变注定无解。但最新研究撕碎了这个认知——90%的医生忽略了一个致命细节:某些突变竟能绕过传统激活路径,直接劫持ERK1/2信号疯狂驱动肿瘤。当所有人聚焦细胞内区域时,这个藏在细胞外的R252C突变却用二硫键"锁死"受体,让常规药物完全失效。更关键的是,第二代抑制剂阿法替尼竟能精准斩断这条暗道,个案中成功控制多发性肿瘤。可为什么同样突变,有人用它起死回生,有人却白白错过黄金期?答案就藏在你从未注意的分子构象里。
— 内容由好学术AI分析文章内容生成,仅供参考。
表皮生长因子受体(EGFR)是肺癌等恶性肿瘤的关键致癌驱动基因。针对其经典突变类型,已有系列靶向药物应用于临床。然而,部分患者携带的EGFR罕见突变亚型,其致癌分子机制阐释不充分,导致临床治疗反应不佳。因此,识别并阐明罕见EGFR突变的致病机制,是当前肿瘤精准医疗领域亟待突破的难点之一。
近日,中国科学院分子细胞科学卓越创新中心等,发现了新的EGFR突变体(EGFR R252C),揭示了其通过直接磷酸化并激活ERK1/2促进肿瘤进展的新功能,并验证了阿法替尼对该突变肿瘤的临床治疗潜力。
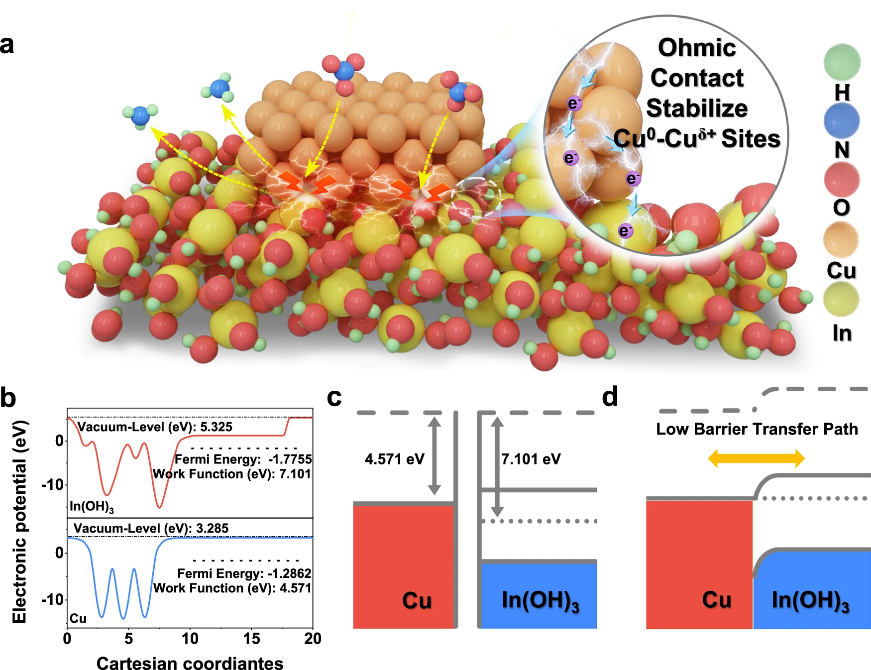
通过基因测序,研究在同时罹患肺癌和脑胶质瘤患者的肿瘤样本中,发现了EGFR罕见突变——位于细胞外区域的第252位精氨酸突变为半胱氨酸,即EGFR R252C。但是,该突变在肿瘤中的具体生物学功能与机制尚不明确。
研究发现,在无配体结合条件下,R252C突变像一把“分子锁”一样,通过新形成的二硫键直接将两个EGFR分子牢牢锁在一起,形成稳定的二聚体。这种异常的二聚化,引发了EGFR受体结构的改变,使得突变后的EGFR本身几乎不发生自磷酸化,却能够绕过经典步骤,直接与下游的关键信号分子ERK1/2结合,磷酸化并激活ERK1/2,促进体内肿瘤细胞增殖和肿瘤生长。药物筛选和分析发现,第二代EGFR酪氨酸激酶抑制剂阿法替尼能够抑制由EGFR R252C突变驱动的ERK1/2活化和肿瘤生长。在针对该患者的个案治疗中,阿法替尼的使用,能够控制其多发性肺癌和胶质瘤的进展,并延长无进展生存期。
该研究从机制层面解释了这一罕见EGFR突变的致癌性,为携带该突变的患者提供了直接可行的治疗选择。
相关研究成果在线发表在《自然-通讯》(Nature Communications)上。研究工作得到科学技术部、国家自然科学基金委员会、中国科学院等的支持。

患者来源的R252C突变对EGFR的替代激活促进癌症进展
© 版权声明
本文由分享者转载或发布,内容仅供学习和交流,版权归原文作者所有。如有侵权,请留言联系更正或删除。







相关文章




之前患者也有罕见EGFR,换药后效果才见起色
这个R252C突变在别的癌种也会出现吗?如果会,治疗方案会不一样吗?
阿法替尼居然能管这么多,太惊喜了👍