







文章导读
你知道吗?电化学界面的电子转移机制可能比我们想象得更精密。南京大学团队最新研究发现,在硝酸根还原制氨反应中,电极极化诱导形成的空轨道竟是启动反应的关键"开关"。通过设计特殊异质结催化剂,他们成功稳定了高活性钴中心,实现了96.7%的氨合成效率,并能稳定运行1000小时以上。这项突破不仅颠覆了传统认知,更为设计高效催化体系提供了全新视角——原来,调控电子自旋态就能构建理想的电子转移通道。
— 内容由好学术AI分析文章内容生成,仅供参考。
电化学界面电子转移机制是设计高效催化中心、提升电能-化学能转换效率的关键理论基础。经典Marcus理论将电解液视为连续介电体,指出反应分子与电解液及电极催化中心的相互作用引发核坐标波动,使界面电子转移遵循Franck-Condon原理与能量守恒定律。然而,该理论未能清晰阐明电子转移过程中的轨道特征。闫世成课题组致力于揭示电化学界面电子转移的微观轨道特征,为高活性催化中心的设计提供物理参数。团队前期研究基于电化学氧化反应,提出了催化中心空轨道作为界面电荷转移通路的新机制(Nat. Commun. 2023, 14, 7987; J. Am. Chem. Soc. 2024, 146, 4814),并发展了利用电极材料热物理效应构建高效界面电荷转移通道的方法,例如:热抑制电荷歧化稳定高价空轨道活性中心(PNAS 2024, 121, e2316054120)、热因瓦效应调控自旋态(Phys. Rev. Lett. 2024, 133, 258001)、以及热应变调控d带中心(Nat. Commun. 2024, 15, 1780)。在最新研究中,课题组以电化学硝酸根还原制氨为模型反应体系,聚焦于界面能量学稳定低自旋Co3+(t2g6eg0)催化活性中心。研究证实,在电化学还原反应界面,电子转移同样遵循空轨道机制。这一发现表明:电极极化诱导形成的空轨道,可能是启动电化学反应的重要标志。
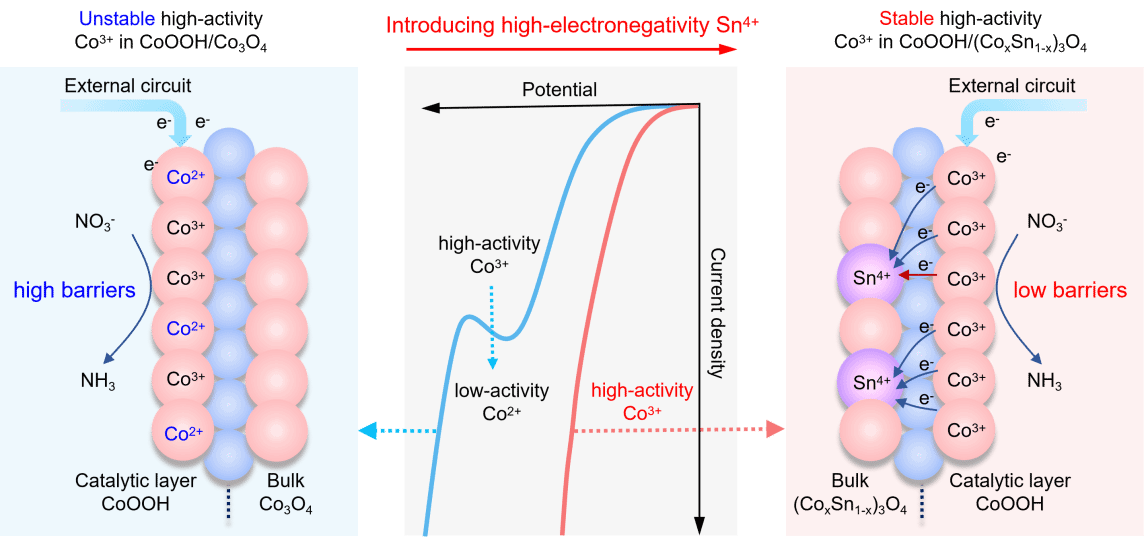
图1. CoOOH/(CoxSn1-x)3O4异质结通过界面电场稳定Co3+的机制示意图。
为揭示电化学硝酸根还原反应(NO3RR)过程中界面电子转移机制,课题组设计了一种CoOOH/(CoxSn1-x)3O4异质结催化剂。其原理在于:通过在Co3O4基底中掺入锡(Sn)元素形成(CoxSn1-x)3O4,增大了其与 CoOOH 催化层之间的功函差异,从而显著增强了异质结界面电场;这一强化的界面电场能够在负电压(还原电位)作用下稳定 CoOOH层中的低自旋 Co3+(t2g6eg0)活性中心。以此稳定、高效的催化活性中心,实现在高电流密度下NO3RR的持久高效催化,并服务于电子转移机制的深入研究。
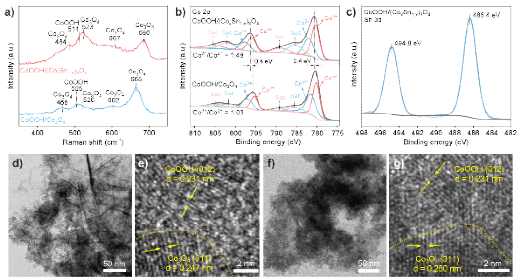
图2. CoOOH/Co3O4和CoOOH/(CoxSn1-x)3O4的结构表征。a) 拉曼光谱;b) Co 2p的XPS 光谱;c) Sn 3d的XPS谱;d)-e) CoOOH/Co3O4的高分辨透射电镜图;f)-g) CoOOH/(CoxSn1-x)3O4的高分辨透射电镜图。
采用电化学沉积法制备了CoOOH/Co3O4和CoOOH/(CoxSn1-x)3O4催化剂。拉曼光谱表征结果证实了尖晶石型Co3O4与CoOOH在催化剂中的共存。XPS 拟合分析表明,相较于未掺杂的CoOOH/Co3O4,Sn掺杂的CoOOH/(CoxSn1-x)3O4中Co3+/Co2+比例显著升高。这一现象归因于Sn4+电负性(1.706)高于Co3+(1.693),表明Sn掺杂有助于稳定高价态的 Co (Co3+)。高分辨透射电镜 (HRTEM) 观察进一步显示,Sn 掺杂后,(CoxSn1-x)3O4的(311) 晶面间距由未掺杂Co3O4的0.247 nm增大至0.250 nm,这种晶格膨胀效应主要源自Sn4+的离子半径(0.069 nm)大于Co3+的离子半径(0.061 nm)。
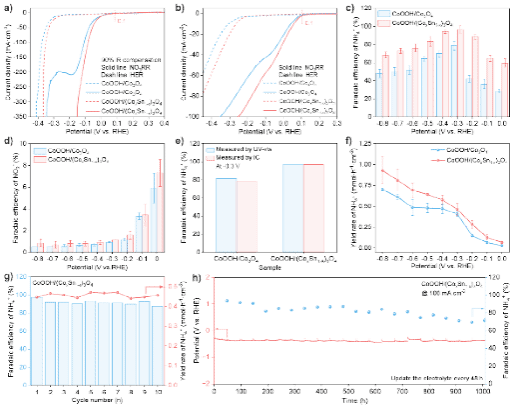
图3. CoOOH/Co3O4和CoOOH/(CoxSn1-x)3O4电催化NO3RR性能表征。a) LSV曲线;b) I-t拟合的LSV曲线;c)-d) NH4+和NO2-的法拉第效率;e) 不同方法测试NH4+法拉第效率对比;f) NH4+产率;g) 循环稳定性测试;h) 恒电流稳定性测试。
在0.1 V (vs. RHE) 电位下,CoOOH/Co3O4和CoOOH/(CoxSn1-x)3O4催化剂均表现出硝酸根还原反应的起始活性。然而,CoOOH/(CoxSn1-x)3O4展现出更优越的本征NO3RR活性及更快的反应动力学。相比之下,CoOOH/Co3O4在反应过程中伴随一个显著的还原峰,该峰归因于高活性的Co3+被还原为低活性的 Co2+。这一还原过程显著增大了反应的过电位。CoOOH/(CoxSn1-x)3催化剂在 -0.3 V (vs. RHE) 时,NH4+的法拉第效率高达96.7%。此外,该催化剂在10次连续循环测试及-100 mA/cm2恒定电流密度下持续运行1000小时后,性能均未出现衰减。综上,CoOOH/(CoxSn1-x)3催化剂在硝酸根还原反应中展现出卓越的催化活性、高选择性和优异的长期稳定性。
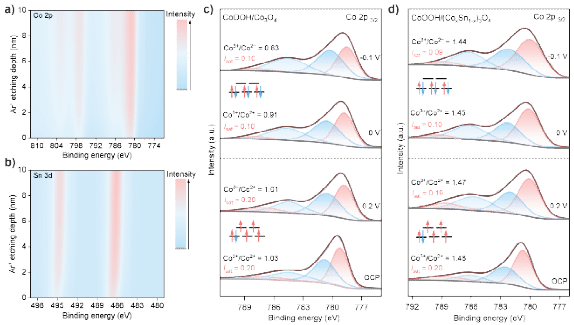
图4. 自旋态转变及活性中心的稳定性。CoOOH/(CoxSn1-x)3O4的a) Co 2p和b) Sn 3d XPS 深度剖析;c) CoOOH/Co3O4和d) CoOOH/(CoxSn1-x)3O4的Co 2p3/2芯能级的原位XPS谱。
XPS深度分析结果显示,在0-10 nm深度范围内,Co和Sn的含量保持稳定,证实了Sn的成功掺杂及其均匀分布。原位XPS进一步表征了电化学反应过程中价态与自旋态的变化:卫星峰强度(Isat)定义为Isat = Asat/(Asat + Aprimary),其中Asat和Aprimary分别为光谱拟合后卫星峰与主峰Co 2p3/2的峰面积。Isat与自旋态呈正相关:Isat越高对应自旋态越高。此外,特征卫星峰位置可区分Co2+与Co3+物种:Co2+在主峰上方4-6 eV处呈现强卫星峰,而Co3+则在结合能高9-10 eV处显示弱卫星峰。开路电位下的Co 2p光谱分析表明,CoOOH/Co3O4和CoOOH/(CoxSn1-x)3O4均为高自旋态,两者Isat值一致(0.2)。但氧化态存在显著差异:Sn掺杂材料的Co3+/Co2+比值更高(1.48,未掺杂材料为1.03),表明Sn掺杂可稳定Co3+物种。将电位从0.2 V调至-0.1 V引发了显著变化,在CoOOH/Co3O4中,Co3+/Co2+比值从1.03显著降至0.83,表明阴极条件下Co3+被还原为活性较低的Co2+;而CoOOH/(CoxSn1-x)3O4的Co3+/Co2+比值保持稳定(仅从1.48微变为1.44),证实Sn掺杂可有效保护活性Co3+中心免受还原。同时,当电位低于0 V时,两种催化剂的Isat值均从0.2降至0.1(0 V),表明Co3+从高自旋态(t2g4eg2,eg轨道单占据)转变为低自旋态(t2g6eg0,eg轨道未占据)。这种自旋态转变至关重要,因为它提供了电子转移所需的未占据轨道,从而启动NO3RR。
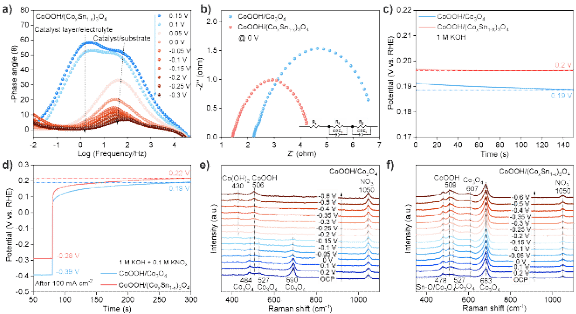
图5. 电子转移及活性中心指认。a) CoOOH/(CoxSn1-x)3O4的原位EIS Bode图;b) 在0 V 时的EIS Nyquist图;c) 开路电压测试;d) 在100 mA/cm2的电流下极化后的开路电压测试;e)CoOOH/Co3O4和f) CoOOH/(CoxSn1-x)3O4的原位拉曼光谱。
电化学阻抗谱分析表明,Sn掺杂显著降低了CoOOH/(CoxSn1-x)3O4的界面电阻。同时,该体系展现出更高的开路电压,这表明其界面结电场更强。进一步地,在-100 mA/cm2极化处理后,CoOOH/(CoxSn1-x)3O4仍能维持较高的开路电压,这暗示其表面活性催化物种具有更高的平均氧化态。原位拉曼光谱结果证实,CoOOH是反应的主要活性相。对于未掺杂的CoOOH/Co3O4,负电压下部分Co3+会被还原为Co(OH)2;而CoOOH/(CoxSn1-x)3O4中,得益于异质结的强界面相互作用,Co3+活性位点展现出更高的稳定性,这直接导致了催化活性与稳定性的增强。
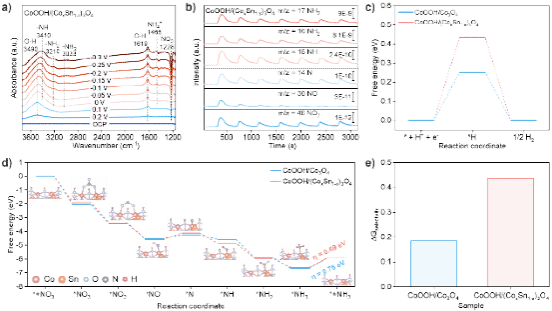
图6. CoOOH/Co3O4和CoOOH/(CoxSn1-x)3O4的电化学反应机制。a) 原位电化学ATR-SEIRAS光谱;b) 电化学在线质谱;c) 氢析出的反应自由能;d) 硝酸根还原反应的自由能;e) 硝酸根还原反应产物选择吉布斯自由能。
结合原位电化学ATR-SEIRAS与电化学在线质谱技术,我们提出了NO3RR的反应路径:NO3- → *NO3 → *NO2 → *NO → *N → *NH → *NH2 → *NH3 → NH3。进一步通过密度泛函理论计算吸附中间体的吉布斯自由能,结果显示CoOOH/(CoxSn1-x)3O4催化剂具有以下优势:其速率决定步骤(即 NH3的脱附)能垒更低,表明其动力学过程更有利;同时,该催化剂还具有较高的氢吸附自由能(不利于析氢副反应),以及对NO3RR更高的反应选择性。这些结果共同证实了CoOOH/(CoxSn1-x)3O4对NO3RR具有优异的选择性。
本研究揭示,空轨道作为关键电子转移通道,在调控电化学反应启动与效率中起核心作用。具体而言,反应启动的关键在于电压驱动下空轨道电子转移通路的构建;对于电化学还原反应,催化中心的自旋态转变正是创建空轨道的有效方式。这一发现为高活性催化中心设计提供了轨道层面的理论依据:其中,调控电子自旋构型转变是催化中心空轨道形成的重要途径,此外,构建界面电场可在高电流密度下稳定空轨道。相关研究成果以“Unlocking Durable and Efficient Nitrate-to-Ammonia Electrocatalysis via Interface-Stabilized Trivalent Cobalt”为题,发表于Angewandte Chemie International Edition(doi.org/10.1002/anie.202522042)。南京大学现代工学院2022级博士生郑倩、2024级硕士生刘泽华与2022级博士生颜元东为该论文的共同第一作者,闫世成教授为通讯作者。研究工作得到邹志刚院士的悉心指导,并获国家自然科学基金、江苏省双碳专项资金等项目资助。
论文信息:
论文题目:Unlocking Durable and Efficient Nitrate-to-Ammonia Electrocatalysis via Interface-Stabilized Trivalent Cobalt
论文作者:Qian Zheng, Zehua Liu, Yuandong Yan, Shicheng Yan*, and Zhigang Zou
论文链接:https://doi.org/10.1002/anie.202522042
© 版权声明
本文由分享者转载或发布,内容仅供学习和交流,版权归原文作者所有。如有侵权,请留言联系更正或删除。





相关文章



电化学小白表示一脸懵,但感觉作者研究得很深入
催更一下,什么时候能看到更详细的实验数据分享?
96.7%的法拉第效率,这数据太强了,实际应用前景怎么样?
看不懂这些图,但能发在Angew上肯定很厉害,南大团队牛啊!
Sn掺杂稳定Co3+这个思路挺巧妙的,之前好像没人这么做过
虽然看不懂那些专业术语,但感觉这个发现对环保应该有帮助吧?🤔
这个空轨道机制解释得挺清楚的,比我之前看的文章更具体👍