电子科技大学医学院张存金教团队授在Journal of Clinical Investigation发表论文







文章导读
多发性硬化为何难以根治?关键病因终于被揭开!电子科技大学医学院张存金教授团队在《Journal of Clinical Investigation》发表重磅研究,首次发现一种名为ANKRD55的蛋白,竟是驱动T细胞异常活化、引发神经炎症的“幕后黑手”。该团队通过单细胞测序与动物模型证实,敲除ANKRD55可显著缓解症状,抑制免疫攻击。更惊人的是,它通过调控CCT分子伴侣复合物,直接影响免疫突触形成——这一发现颠覆了对T细胞活化机制的认知。这项突破不仅揭示了多发性硬化急性期的核心病理,更为开发靶向新药提供了关键线索,或将改写20年来无新疗法的局面。
— 内容由好学术AI分析文章内容生成,仅供参考。
多发性硬化(MS)是一种自身免疫性中枢神经系统疾病,病程复杂且缺乏根治手段。T细胞的异常活化和炎症反应被认为是MS发病的核心机制,但是T细胞如何被初始活化,仍然很不清楚,多发性硬化中枢急性期的神经炎症治疗在近20年缺乏临床新疗法。
针对上述临床急性期难题,近日医学院张存金教授以最后通讯作者在Journal of Clinical Investigation(IF=13.6,双1区)上发表了题为 “ANKRD55 is a key regulator of T cell inflammation in multiple sclerosis”的研究论文。这项工作揭示了ANKRD55是多发性硬化急性期T细胞炎症活化的关键,为MS等自身免疫性疾病的治疗提供了新的潜在靶点。医学院博士研究生吴楚钰、硕士研究生杨雪以及省医院姜梅玲老师为论文共同第一作者,医学院张存金教授、王晨辉教授及中国药科大学庞涛研究员为本文的通讯作者。

该研究通过对临床MS患者外周血和脑脊液的单细胞转录组分析,首次发现一种功能未知的蛋白ANKRD55,其主要表达于活化的炎性CD4⁺ T细胞中,且在MS患者中枢显著上调,提示ANKRD55参与急性期异常免疫激活。
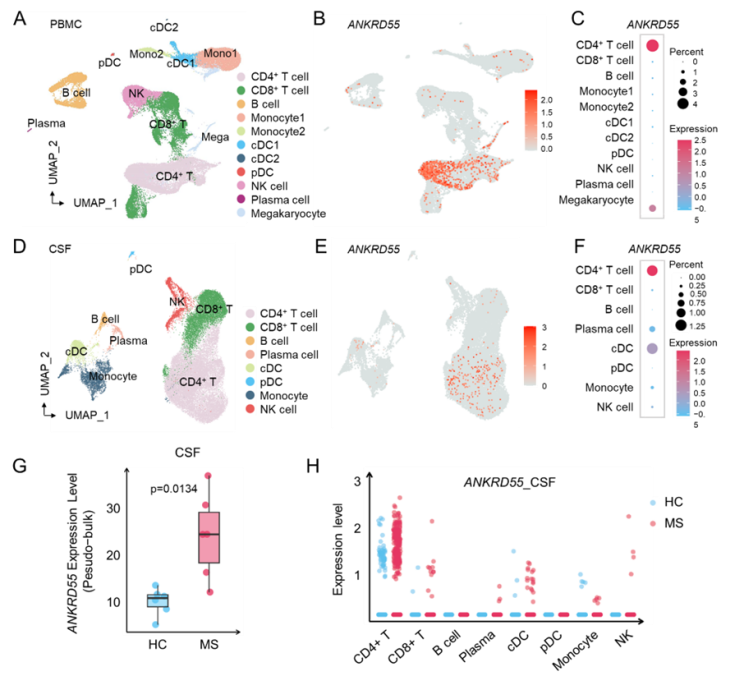
通过构建ANKRD55敲除小鼠,并诱导EAE模型(MS 动物模型),该研究发现敲除ANKRD55疾病严重程度明显降低。且敲除ANKRD55抑制了CD4+T细胞的活化、增殖,以及其向TH1,TH17的分化。进一步通过转录组测序发现ANKRD55敲除显著下调了TCR受体信号通路。同时,通过免疫共沉淀联合质谱发现ANKRD55和CCT复合物的多个亚基发生相互作用。CCT是一个分子伴侣,作用是帮助tubulin,actin等骨架蛋白的折叠。在免疫突触过程中CCT 复合物在中心体中控制TCR诱导的微管蛋白的极化,从而控制T细胞的活化和极性。研究团队发现ANKRD55 和CCT 在免疫突触过程中在中心体共定位。ANKRD55在CD4+T细胞中通过和CCT5竞争与CCT1/3/6结合帮助CCT复合物正确组装调控免疫突触和TCR进而促进MS中的T细胞炎症。抑制ANKRD55炎症的检查点抑制剂,可显著改善抑制疾病进展,提示其有望作为急性期的创新治疗策略。

该研究揭示了ANKRD55在多发性硬化中的关键作用,揭示了其对T细胞炎症反应中的核心调控作用,为MS等自身免疫性疾病提供新的潜在治疗新策略。
原文链接:https://www.jci.org/articles/view/195214
© 版权声明
本文由分享者转载或发布,内容仅供学习和交流,版权归原文作者所有。如有侵权,请留言联系更正或删除。





相关文章
![上海交大药学院吴华团队发文:去芳构化 [3,3]-σ重排策略实现喹啉C3-H巯基化](https://oss.hxs123.cn/wp-content/uploads/2026/03/60618f855c7375682b139c07ae418930.jpg)




暂无评论...