科学家开发出高通量单细胞基因组和转录组联合测序方法







文章导读
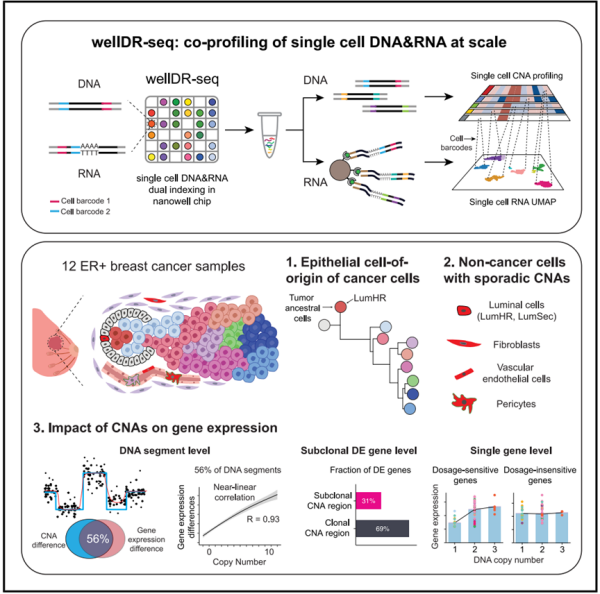
乳腺癌的起源细胞究竟是什么?中国科学院科学家开发出革命性高通量单细胞基因组和转录组联合测序方法wellDR-seq,首次在单细胞水平破解ER阳性乳腺癌的起源之谜。通过对12例样本中超过3万个单细胞的双组学解析,研究鉴定出肿瘤起始祖先亚克隆源自LumHR细胞,并定量揭示基因拷贝数变化与表达变化的复杂关系:56%的CNAs与表达正相关,区分出“剂量敏感性”基因(如PGR)和“剂量不敏感性”基因(如PIK3CA)。这项发表于《细胞》杂志的突破,不仅为乳腺癌早期诊断、预后判断和精准治疗提供新依据,还开创了肿瘤演化和基因型-表型研究的通用技术路径。
— 内容由好学术AI分析文章内容生成,仅供参考。
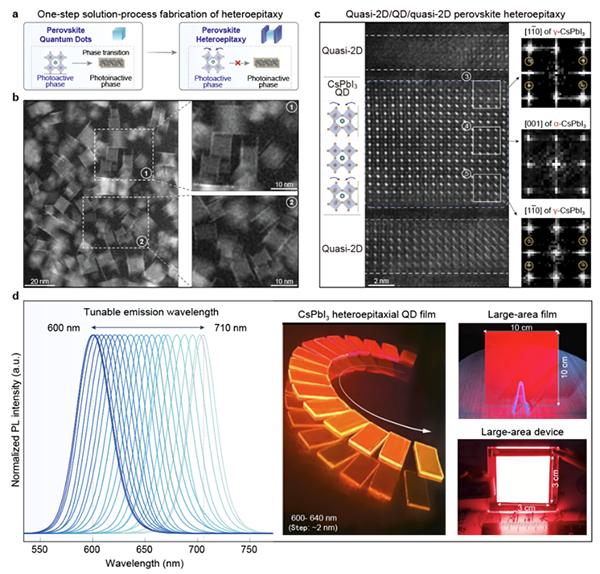
近日,中国科学院分子细胞科学卓越创新中心等,开发出高通量单细胞基因组和转录组联合测序方法wellDR-seq,对12例乳腺癌中超过3万个单细胞进行双组学解析。
研究团队鉴定并表征ER阳性乳腺癌肿瘤起始祖先亚克隆,首次在单细胞水平上,定量描述基因拷贝数变化与基因表达变化之间的复杂剂量调控关系。该研究为乳腺癌的早期诊断、预后判断和精准治疗,提供了新的理论依据和技术路线。
乳腺癌是常见的恶性肿瘤之一。乳腺癌有多种亚型,而人的正常乳腺有三种上皮细胞。不同亚型的乳腺癌由何种正常上皮细胞演化尚不清楚。鉴定不同乳腺癌的起源,以及导致后续乳腺癌增殖和演化的基因,是攻克乳腺癌的关键。
但是,祖先克隆群体存在比例低、突变少,只有通过高通量、高基因组分辨率的单细胞基因组和转录组共测序技术,才能够鉴定该群体,并获得该群体的表型,这在技术上颇具挑战性。
科研团队开发出高通量、高基因组分辨率单细胞DNA与RNA联合测序方法——wellDR-seq。该方法对DNA和RNA的生化反应进行反复优化,并结合双重索引标记技术和纳升小孔芯片技术,首次实现高精度地同时对数千个单细胞基因组和转录组的联合测序解析。
科研团队对MDA-MB-231细胞系的双组学数据分析发现,仅通过scRNA-seq数据推断的拷贝数信息不可靠。这显示出仅依靠单细胞转录组分析DNA克隆结构的局限性,强调了直接对DNA和RNA进行联合测序在揭示肿瘤真实演化路径中的重要性。
科研团队还在4名ER阳性患者中识别出肿瘤的祖先亚克隆。这些祖先亚克隆携带少量拷贝数变异(CNAs),进而在肿瘤进展过程中获取更多CNAs并大规模扩增,最终形成主要肿瘤细胞群体。
科研团队通过wellDR-seq发现,这些祖先细胞都源自LumHR细胞谱系,表明LumHR细胞是ER阳性乳腺癌的起源细胞。
研究还发现,非癌性细胞存在偶发CNAs。这些CNAs存在于上皮细胞中,也存在于基质细胞中。在上皮细胞中,这些突变细胞主要存在于两种腔面型细胞中,而未在MyoEpi中发现,这些突变主要发生在常染色体上。存在于基质细胞中的突变主要发生在X染色体上。这提示,腔面型上皮更易导致肿瘤发生,为肿瘤发生的早期预警提供了新思路。
该研究首次在单细胞层面量化了基因拷贝数变化对基因表达的影响。研究发现,在染色体片段水平,56%CNAs与基因表达变化正相关。在这56%的CNAs中,基因表达量增加与拷贝数增加的相关性是接近线性的。研究比较肿瘤亚克隆间的差异表达基因发现,这些基因多数定位于拷贝数无变化的区域,而非拷贝数变化的区域。
研究还发现,在单个基因层面上,拷贝数变化和基因表达变化存在较大差异。研究团队将基因分为“剂量敏感性”基因和“剂量不敏感性”基因。
“剂量敏感性”基因如PGR、AURKA和RB1,这些基因的表达水平与拷贝数变化相关,其表达量随基因拷贝数增减而同步变化。“剂量不敏感性”基因如PIK3CA、BRCA1和TP53,这些基因的表达水平与拷贝数变化无关,即使基因拷贝数发生变异,其表达水平仍保持稳定。
这为理解肿瘤的遗传学和功能学提供了新视角,解释了为何某些基因突变或拷贝数变异会强烈驱动肿瘤进展,而另一些则不然。
上述研究解析了乳腺癌起源,定量表征了基因剂量效应。同时,wellDR-seq是通用技术,可被广泛应用于不同肿瘤的起源和演化、基因型与表型的互作等研究。
9月4日,相关研究成果在线发表在《细胞》(Cell)上。
wellDR-seq实现乳腺癌样本的高通量单细胞全基因组与转录组的联合分析
© 版权声明
本文由分享者转载或发布,内容仅供学习和交流,版权归原文作者所有。如有侵权,请留言联系更正或删除。





相关文章



暂无评论...